The spreading epidemic of allergies and asthma has heightened interest in IgE, the central player in the allergic response. The activity of IgE is associated with a network of proteins; prominent among these are its two principal receptors, Fc RI (high-affinity Fc receptor for IgE) and CD23, as well as galectin-3 and several co-receptors for CD23, notably CD21 and various integrins. Here, we review recent progress in uncovering the structures of these proteins and their complexes, and in our understanding of how IgE exerts its effects and how its expression is regulated. The information that has emerged suggests new therapeutic directions for combating allergic disease.
RI (high-affinity Fc receptor for IgE) and CD23, as well as galectin-3 and several co-receptors for CD23, notably CD21 and various integrins. Here, we review recent progress in uncovering the structures of these proteins and their complexes, and in our understanding of how IgE exerts its effects and how its expression is regulated. The information that has emerged suggests new therapeutic directions for combating allergic disease.
RI (high-affinity Fc receptor for IgE) and CD23, as well as galectin-3 and several co-receptors for CD23, notably CD21 and various integrins. Here, we review recent progress in uncovering the structures of these proteins and their complexes, and in our understanding of how IgE exerts its effects and how its expression is regulated. The information that has emerged suggests new therapeutic directions for combating allergic disease.Summary
- Remarkable progress has been made in recent years on the structural determination of proteins in the IgE network and on the functions and regulation of IgE. This is beginning to feed into IgE-targeted therapies for allergy.
- The shape of the IgE molecule differs dramatically from that of IgG. X-ray and nuclear magnetic resonance studies have also revealed conformational changes that occur on IgE binding to its high-affinity receptor FcRI (high-affinity Fc receptor for IgE) on mast cells and antigen-presenting cells, events that lead, respectively, to sensitization (and the immediate hypersensitivity reaction) and the facilitation of allergen presentation.
- The structural data also provide clues to the unique nature of the high-affinity IgE–FcRI interaction, and indicate possibilities for blocking the interaction; validation of IgE as a target is demonstrated by the success of the IgE-specific monoclonal antibody omalizumab in the treatment of asthma.
- The presence of an unusually long extracellular membrane-proximal domain in membrane IgE may also determine its ability to act as an antigen receptor on B cells and to respond to particular antigens (allergens).
- The trimeric structure of the C-type lectin, low-affinity IgE receptor CD23, and its susceptibility to cleavage by ADAM10 (a disintegrin and metalloproteinase 10) at the cell surface, provide important clues to the mechanism of IgE homeostasis.
- CD23 has multiple ligands, including IgE, CD21 and various integrins, enabling it to carry out several other functions, including IgE-dependent antigen presentation and cellular cytotoxicity.
- IgE is transported to mucosal tissues by CD23 and is also synthesized by the resident B cells. Its concentration is maintained in the tissue by the number of mast cells that express FcRI at high levels and the slow rate of dissociation of IgE from FcRI.
- Class switching to IgE and affinity maturation of the antibodies occur in mucosal tissues, and this may limit the ability of IgE antibodies to mediate systemic anaphylaxis.
- Various mechanisms contrive to suppress the production of IgE synthesis to tolerable levels, as well as limiting its anatomical distribution. Some mechanisms operate at the level of class-switch recombination, others at the level of survival of the IgE-switched cells.
- IgE transport in both directions through the gastrointestinal epithelium may be involved in early sensitization to allergens. Studies of the mechanism of early sensitization may suggest means of preventing the development of allergic disease.
- Small molecule inhibitors of the IgE–FcRI interaction may supersede IgE-specific antibodies, but the combination of this approach with immunotherapy may be required for more effective therapeutic intervention in allergy and asthma.
Forty years ago, in February 1968, the name of the fifth and final class of human antibody, IgE, was officially adopted at a meeting under the auspices of the World Health Organization Immunoglobulin Reference Laboratory, held in Lucerne, Switzerland. The year before this, a series of papers had been published marking the culmination of studies by the Ishizakas in the USA, Bennich and Johansson in Sweden, and Humphrey and Stanworth in the UK. These established unequivocally that this new class of antibody was the factor capable of transferring sensitivity to allergens. The history of the discovery of IgE has been related by Stanworth1.
IgE is thought to have evolved in mammals as the first line of defence against pathogens2. But modern civilization has engendered a large increase in the cost:benefit ratio of this mechanism, such that a large and increasing proportion of the population — now around one in three — suffers from allergies3. It has emerged that IgE acts as part of a protein network4, which includes its two principal receptors, FcRI (high-affinity Fc receptor for IgE) and CD23, as well as the IgE- and FcRI-binding protein galectin-3. In addition, the function of CD23 is extended by several co-receptors. These comprise the complement receptors CD21 (also known as CR2),  M
M 2-integrin (otherwise known as CD18/CD11b or CR3) and X2-integrin (also known as CD18/CD11c or CR4); the vitronectin receptor (also known as V3-integrin); and V5-integrin. Here, we review the remarkable progress of the past several years on the structural determination of these proteins and of the complexes formed between them, and on the functions and regulation of IgE in the context of the extended protein network. The success of the IgE-specific antibody, omalizumab (Xolair; Novartis Pharmaceuticals Ltd), in the treatment of asthma and other allergic diseases demonstrates the virtues of targeting IgE for therapy5. We expect that a deeper understanding of the IgE network will lead to a new generation of IgE-targeted therapies.
2-integrin (otherwise known as CD18/CD11b or CR3) and X2-integrin (also known as CD18/CD11c or CR4); the vitronectin receptor (also known as V3-integrin); and V5-integrin. Here, we review the remarkable progress of the past several years on the structural determination of these proteins and of the complexes formed between them, and on the functions and regulation of IgE in the context of the extended protein network. The success of the IgE-specific antibody, omalizumab (Xolair; Novartis Pharmaceuticals Ltd), in the treatment of asthma and other allergic diseases demonstrates the virtues of targeting IgE for therapy5. We expect that a deeper understanding of the IgE network will lead to a new generation of IgE-targeted therapies.
RI (high-affinity Fc receptor for IgE) and CD23, as well as the IgE- and FcRI-binding protein galectin-3. In addition, the function of CD23 is extended by several co-receptors. These comprise the complement receptors CD21 (also known as CR2), M2-integrin (otherwise known as CD18/CD11b or CR3) and X2-integrin (also known as CD18/CD11c or CR4); the vitronectin receptor (also known as V3-integrin); and V5-integrin. Here, we review the remarkable progress of the past several years on the structural determination of these proteins and of the complexes formed between them, and on the functions and regulation of IgE in the context of the extended protein network. The success of the IgE-specific antibody, omalizumab (Xolair; Novartis Pharmaceuticals Ltd), in the treatment of asthma and other allergic diseases demonstrates the virtues of targeting IgE for therapy5. We expect that a deeper understanding of the IgE network will lead to a new generation of IgE-targeted therapies.Structures and interactions in the IgE network
The IgE molecule. IgE shares the same basic molecular architecture as antibodies of other classes, with two identical heavy chains and two identical light chains, but, as illustrated in Fig. 1, the heavy -chain contains one more domain than the heavy  -chain of IgG. The pair of C3 and C4 domains are homologous in sequence, and similar in quaternary structure, to the pair of C2 and C3 domains of IgG, so that it is the pair of C2 domains, located in the position equivalent to that occupied by the flexible hinge region of IgG, that is the most obvious distinguishing feature of IgE. Initially it was thought that this 'extra' domain pair might simply act as a spacer between the antigen-binding Fab 'arms' and the C3–C4 part of the Fc region (Fc3-4), but elegant studies using fluorescence resonance energy transfer (FRET) determined that the distance between the N- and C-termini of the molecule was less than half that expected for an extended Y-shaped structure (6.9 Å compared with 17.5 Å)6; furthermore, this was also true for the IgE molecule bound to FcRI (Refs 6, 7).
-chain of IgG. The pair of C3 and C4 domains are homologous in sequence, and similar in quaternary structure, to the pair of C2 and C3 domains of IgG, so that it is the pair of C2 domains, located in the position equivalent to that occupied by the flexible hinge region of IgG, that is the most obvious distinguishing feature of IgE. Initially it was thought that this 'extra' domain pair might simply act as a spacer between the antigen-binding Fab 'arms' and the C3–C4 part of the Fc region (Fc3-4), but elegant studies using fluorescence resonance energy transfer (FRET) determined that the distance between the N- and C-termini of the molecule was less than half that expected for an extended Y-shaped structure (6.9 Å compared with 17.5 Å)6; furthermore, this was also true for the IgE molecule bound to FcRI (Refs 6, 7).
-chain contains one more domain than the heavy -chain of IgG. The pair of C3 and C4 domains are homologous in sequence, and similar in quaternary structure, to the pair of C2 and C3 domains of IgG, so that it is the pair of C2 domains, located in the position equivalent to that occupied by the flexible hinge region of IgG, that is the most obvious distinguishing feature of IgE. Initially it was thought that this 'extra' domain pair might simply act as a spacer between the antigen-binding Fab 'arms' and the C3–C4 part of the Fc region (Fc3-4), but elegant studies using fluorescence resonance energy transfer (FRET) determined that the distance between the N- and C-termini of the molecule was less than half that expected for an extended Y-shaped structure (6.9 Å compared with 17.5 Å)6; furthermore, this was also true for the IgE molecule bound to FcRI (Refs 6, 7). |
| FIGURE 1 | The domain structures of IgE and IgG.
Schematic representations of the polypeptide and domain structures of human IgE and IgG1, showing the intra- and inter-domain disulphide bridges, and the sites of N-linked glycosylation.
|
When the crystal structure of the complete IgE-Fc, including the C2 domains, was determined, the molecule was found to be even more acutely bent than had been anticipated, with the C2 domains folded back and making extensive contact with the C3 domains, and even touching the C4 domains8 (Fig. 2a, b). The structure was entirely consistent with X-ray and neutron scattering data obtained in solution9, showing that this compact, bent conformation was indeed its native structure, and was markedly different from the extended and flexible structure of IgG7. Nevertheless, within this overall compact structure, IgE displays considerable conformational flexibility both in tertiary (intra-domain) and quaternary (inter-domain) structure. Comparison of crystal structures of Fc3-4 alone and in complex with FcRI (Ref. 10) (see below) show that the C3 domains can adopt 'closed' (Fig. 2c) or 'open' (Fig. 2d) conformations by rotating relative to C4. The asymmetrically bent IgE-Fc (Fig. 2a, b), with its C2 domains packed against the two C3 domains, has one C3 domain in the 'open' conformation, while the other is 'closed', and this structural asymmetry explains a key feature of the interaction with FcRI, as we discuss later.
2 domains, was determined, the molecule was found to be even more acutely bent than had been anticipated, with the C2 domains folded back and making extensive contact with the C3 domains, and even touching the C4 domains8 (Fig. 2a, b). The structure was entirely consistent with X-ray and neutron scattering data obtained in solution9, showing that this compact, bent conformation was indeed its native structure, and was markedly different from the extended and flexible structure of IgG7. Nevertheless, within this overall compact structure, IgE displays considerable conformational flexibility both in tertiary (intra-domain) and quaternary (inter-domain) structure. Comparison of crystal structures of Fc3-4 alone and in complex with FcRI (Ref. 10) (see below) show that the C3 domains can adopt 'closed' (Fig. 2c) or 'open' (Fig. 2d) conformations by rotating relative to C4. The asymmetrically bent IgE-Fc (Fig. 2a, b), with its C2 domains packed against the two C3 domains, has one C3 domain in the 'open' conformation, while the other is 'closed', and this structural asymmetry explains a key feature of the interaction with FcRI, as we discuss later. |
| FIGURE 2 | The structures of IgE-Fc fragments.
a, b | Two (approximately orthogonal) views of the complete IgE-Fc (uncomplexed) including the C
2 domains (Protein Data Bank (PDB) ID: 1O0V). The two -chains are coloured purple and green, and the two points of connection to the Fab regions can be seen most clearly in a and b respectively, indicating the extent of the bend in the IgE molecule. c | The structure of the Fc3-4 fragment (uncomplexed) showing the two C3 domains in the 'closed' conformation (PDB ID: 1FP5). d | The structure of the Fc3-4 fragment taken from the crystal structure of the complex of Fc3-4 and the FcRI -chain, showing the C3 domains in the 'open' conformation (PDB ID: 1F6A). |
These structural features of IgE-Fc must also apply to the membrane form of the IgE molecule, which together with the signalling chains Ig and Ig constitutes the receptor for antigen on B cells committed to IgE synthesis. Each heavy chain of all membrane IgE molecules contains an additional extracellular membrane-proximal domain (EMPD) between the C4 domain and the transmembrane sequence, although there are at least two isoforms with EMPDs of different length, short (14 amino acids) and long (66 amino acids)11. Nothing is known of the three-dimensional structure of the EMPDs, which contain inter--chain disulphide bridges, nor the way in which IgE and its Fab regions are presented at the cell surface, but the two isoforms associate with different glycoforms of Ig and, together with other accessory proteins, form functionally distinct antigen receptors12. Whatever the position of the IgE molecule in this complex, the binding site for FcRI is accessible to receptors expressed on another cell surface; the implications of this interaction for allergic disease have yet to be explored13.
and Ig constitutes the receptor for antigen on B cells committed to IgE synthesis. Each heavy chain of all membrane IgE molecules contains an additional extracellular membrane-proximal domain (EMPD) between the C4 domain and the transmembrane sequence, although there are at least two isoforms with EMPDs of different length, short (14 amino acids) and long (66 amino acids)11. Nothing is known of the three-dimensional structure of the EMPDs, which contain inter--chain disulphide bridges, nor the way in which IgE and its Fab regions are presented at the cell surface, but the two isoforms associate with different glycoforms of Ig and, together with other accessory proteins, form functionally distinct antigen receptors12. Whatever the position of the IgE molecule in this complex, the binding site for FcRI is accessible to receptors expressed on another cell surface; the implications of this interaction for allergic disease have yet to be explored13.
The high-affinity receptor Fc RI and its interaction with IgE. FcRI is expressed as an 2 tetramer on mast cells and basophils, and as an 2 trimer on human but not mouse antigen-presenting cells (APCs), monocytes, eosinophils, platelets and smooth-muscle cells, but its distribution is even wider than this14, 15. The two extracellular domains of the -chain (sFcRI) contain the IgE binding function, whereas the signalling motifs (immunoreceptor tyrosine-based activation motifs (ITAMs)) are located in the intracellular sequences of the - and -chains. Although the quaternary arrangement of the chains is not known, X-ray analyses of sFcRI in several different crystal forms have revealed that the two domains are folded back on each other exposing a hydrophobic 'ridge' (Fig. 3a) that was subsequently found to be the IgE binding site. Part of this ridge is formed by the CC' loop of the 2 domain, a segment that strikingly is the only region to exhibit conformational variation among the different unliganded sFcRI structures16, 17.
RI and its interaction with IgE. FcRI is expressed as an 2 tetramer on mast cells and basophils, and as an 2 trimer on human but not mouse antigen-presenting cells (APCs), monocytes, eosinophils, platelets and smooth-muscle cells, but its distribution is even wider than this14, 15. The two extracellular domains of the -chain (sFcRI) contain the IgE binding function, whereas the signalling motifs (immunoreceptor tyrosine-based activation motifs (ITAMs)) are located in the intracellular sequences of the - and -chains. Although the quaternary arrangement of the chains is not known, X-ray analyses of sFcRI in several different crystal forms have revealed that the two domains are folded back on each other exposing a hydrophobic 'ridge' (Fig. 3a) that was subsequently found to be the IgE binding site. Part of this ridge is formed by the CC' loop of the 2 domain, a segment that strikingly is the only region to exhibit conformational variation among the different unliganded sFcRI structures16, 17.
RI and its interaction with IgE. FcRI is expressed as an 2 tetramer on mast cells and basophils, and as an 2 trimer on human but not mouse antigen-presenting cells (APCs), monocytes, eosinophils, platelets and smooth-muscle cells, but its distribution is even wider than this14, 15. The two extracellular domains of the -chain (sFcRI) contain the IgE binding function, whereas the signalling motifs (immunoreceptor tyrosine-based activation motifs (ITAMs)) are located in the intracellular sequences of the - and -chains. Although the quaternary arrangement of the chains is not known, X-ray analyses of sFcRI in several different crystal forms have revealed that the two domains are folded back on each other exposing a hydrophobic 'ridge' (Fig. 3a) that was subsequently found to be the IgE binding site. Part of this ridge is formed by the CC' loop of the 2 domain, a segment that strikingly is the only region to exhibit conformational variation among the different unliganded sFcRI structures16, 17. |
FIGURE 3 | The structure of the Fc RI RI  -chain and its complex with IgE. -chain and its complex with IgE.
a | The structure of the extracellular domains of the Fc
RI -chain (Protein Data Bank (PDB) ID: 1F6A), taken from the crystal structure of the Fc3-4–FcRI -chain complex, with the superimposed structure of free FcRI -chain (PDB ID: 1J87) showing only the region (shown in red) in which the structures differ. This is the CC' loop region, which displays conformational flexibility even within uncomplexed structures determined in different crystal forms. The structural change involves the edge -strand moving from one face of the immunoglobulin fold to the other. b | The structure of the high-affinity complex between Fc3-4 and the extracellular domains of the FcRI -chain, showing the extensive interaction surface and engagement of both C3 domains in the 'open' conformation (PDB ID: 1F6A). The connection to the membrane is at the C-terminal end of the 2 domain. c | Schematic representation of the entire IgE molecule bound to the extracellular domains of the FcRI -chain, according to the structural information from the Fc3-4 complex and the bent IgE-Fc structure. The - and -chains of FcRI, with their immunoreceptor tyrosine-based activation motifs (ITAMs), are also shown. |
The first view of the complex of FcRI with IgE came from the Fc3-4–sFcRI structure18, which showed how the two C3 domains of IgE open up and how each contributes a sub-site of predominantly hydrophobic interaction surface (Fig. 3b). The location of this binding site clearly accounts for the observed 1:1 stoichiometry despite the presence of two identical -chains, and the extensive hydrophobic buried surface explains the high affinity (Ka  1010 M-1). In IgE (or IgE-Fc) with its C2 domains, one of the C3 domains is already in an open configuration and therefore accessible for receptor binding, as described above, whereas the other is not. The conformational changes that are necessary to allow IgE engagement using both sub-sites must therefore also include the C2 domains, and the effect of their presence on the kinetics of the interaction — a reduction in both the on-rate and off-rate of IgE binding — have been quantified19, 20. The kinetics are biphasic19, consistent with the involvement of two sub-sites. It may be necessary for these extensive structural changes in IgE, and the CC' loop of the receptor, to occur in a concerted way to allow IgE disengagement. This would account in part for the exceptionally slow dissociation rate compared to IgG from its structurally homologous receptors20.
1010 M-1). In IgE (or IgE-Fc) with its C2 domains, one of the C3 domains is already in an open configuration and therefore accessible for receptor binding, as described above, whereas the other is not. The conformational changes that are necessary to allow IgE engagement using both sub-sites must therefore also include the C2 domains, and the effect of their presence on the kinetics of the interaction — a reduction in both the on-rate and off-rate of IgE binding — have been quantified19, 20. The kinetics are biphasic19, consistent with the involvement of two sub-sites. It may be necessary for these extensive structural changes in IgE, and the CC' loop of the receptor, to occur in a concerted way to allow IgE disengagement. This would account in part for the exceptionally slow dissociation rate compared to IgG from its structurally homologous receptors20.
RI with IgE came from the Fc3-4–sFcRI structure18, which showed how the two C3 domains of IgE open up and how each contributes a sub-site of predominantly hydrophobic interaction surface (Fig. 3b). The location of this binding site clearly accounts for the observed 1:1 stoichiometry despite the presence of two identical -chains, and the extensive hydrophobic buried surface explains the high affinity (Ka 1010 M-1). In IgE (or IgE-Fc) with its C2 domains, one of the C3 domains is already in an open configuration and therefore accessible for receptor binding, as described above, whereas the other is not. The conformational changes that are necessary to allow IgE engagement using both sub-sites must therefore also include the C2 domains, and the effect of their presence on the kinetics of the interaction — a reduction in both the on-rate and off-rate of IgE binding — have been quantified19, 20. The kinetics are biphasic19, consistent with the involvement of two sub-sites. It may be necessary for these extensive structural changes in IgE, and the CC' loop of the receptor, to occur in a concerted way to allow IgE disengagement. This would account in part for the exceptionally slow dissociation rate compared to IgG from its structurally homologous receptors20.
The crystal structure of the Fc3-4 complex, the acute bend in the IgE-Fc, and the fact that there is very little flexibility expected between the N-terminal end of the C2 domains and the Fab regions, leads to a picture of the receptor-bound IgE molecule (Fig. 3c) that is remarkably similar to that drawn 16 years ago based on the FRET studies7. Therefore, it is likely that there are orientational constraints on the ligands, and location of allergenic epitopes that can effectively crosslink two or more receptor-bound IgE molecules and trigger cell activation. These effects are currently under investigation21. However, the first crystal structure of an allergen–IgE complex, that of the dimeric -lactoglobulin bound by two Fab regions of IgE, shows how the relative position of the two epitopes in this case enables crosslinking to occur22.
3-4 complex, the acute bend in the IgE-Fc, and the fact that there is very little flexibility expected between the N-terminal end of the C2 domains and the Fab regions, leads to a picture of the receptor-bound IgE molecule (Fig. 3c) that is remarkably similar to that drawn 16 years ago based on the FRET studies7. Therefore, it is likely that there are orientational constraints on the ligands, and location of allergenic epitopes that can effectively crosslink two or more receptor-bound IgE molecules and trigger cell activation. These effects are currently under investigation21. However, the first crystal structure of an allergen–IgE complex, that of the dimeric -lactoglobulin bound by two Fab regions of IgE, shows how the relative position of the two epitopes in this case enables crosslinking to occur22.
The low-affinity receptor CD23, its soluble fragments and IgE interactions. CD23 is distinguished structurally from almost all other immunoglobulin receptors as it belongs to the C-type (calcium dependent) lectin superfamily. In the membrane-bound form of CD23, three lectin domain 'heads' are spaced from the membrane by a triple -helical coiled-coil 'stalk' (Fig. 4a). However, the stalk region is susceptible to proteolysis, leading to the release of various soluble fragments (soluble CD23) that have activities that depend on their oligomeric state. The principal endogenous protease that releases soluble CD23 has recently been identified as ADAM10 (a disintegrin and metalloproteinase 10)23, 24. Subsequently, smaller soluble fragments of CD23 are generated of various sizes; those containing stalk sequences form trimers in a concentration-dependent manner or on IgE binding, whereas those lacking any stalk region are monomeric25. In addition to promoting oligomerization, however, the stalk region probably has other functions, as a peptide at the base of the stalk region has been implicated in binding to MHC class II molecules26. In the human form, but not the mouse form, the lectin head domains of CD23 additionally have a C-terminal 'tail' sequence (Fig. 4a). IgE-binding activity is contained within the head, but the name 'low affinity' is a misnomer; although the affinity of a single head for IgE-Fc is low (Ka 106–107M-1), the avidity effect achieved with the trimer leads to an affinity (Ka 108–109M-1) approaching the high-affinity of FcRI (Refs 25, 27).
-helical coiled-coil 'stalk' (Fig. 4a). However, the stalk region is susceptible to proteolysis, leading to the release of various soluble fragments (soluble CD23) that have activities that depend on their oligomeric state. The principal endogenous protease that releases soluble CD23 has recently been identified as ADAM10 (a disintegrin and metalloproteinase 10)23, 24. Subsequently, smaller soluble fragments of CD23 are generated of various sizes; those containing stalk sequences form trimers in a concentration-dependent manner or on IgE binding, whereas those lacking any stalk region are monomeric25. In addition to promoting oligomerization, however, the stalk region probably has other functions, as a peptide at the base of the stalk region has been implicated in binding to MHC class II molecules26. In the human form, but not the mouse form, the lectin head domains of CD23 additionally have a C-terminal 'tail' sequence (Fig. 4a). IgE-binding activity is contained within the head, but the name 'low affinity' is a misnomer; although the affinity of a single head for IgE-Fc is low (Ka 106–107M-1), the avidity effect achieved with the trimer leads to an affinity (Ka 108–109M-1) approaching the high-affinity of FcRI (Refs 25, 27). |
FIGURE 4 | The structure of CD23.
a | A schematic representation of membrane-bound CD23, showing the extracellular trimeric
-helical coiled-coil 'stalk', the three C-type lectin domain 'heads' and the C-terminal 'tails'. N-linked glycosylation sites near the base of the stalk are also shown. b | The three-dimensional structure of one of the head domains. The NMR structure (shown in blue; Protein Data Bank (PDB) ID: 1T8D) and X-ray crystal structures (PDB ID: 2H2T) are superimposed, and the latter is displayed only in the regions where the two structures differ most (shown in red); other differences occur in the N- and C-terminal regions of the domain. The single Ca2+ ion observed in each structure is indicated as a sphere in its respective colour. Binding regions are indicated for: IgE-Fc (C3 domain); CD21 (domains 1 and 2) in a region adjacent to the C-terminal tail, most of which is either absent (NMR structure) or disordered (X-ray structure); and V5-integrin, in the N-terminal region close to the connection to the stalk. |
The short (N-terminal) intracellular sequence of CD23 exists in two splice variants that differ in their first seven (CD23a) or six (CD23b) amino-acid residues. They are differentially expressed: CD23a is expressed by antigen-activated B cells before differentiation into antibody-secreting plasma cells, whereas CD23b expression is induced by interleukin-4 (IL-4) on a variety of inflammatory cells, B cells and, importantly, epithelial cells. The region that differs between the two splice forms accounts for their distinct activities, and the crucial residues within this region have been identified28, 29.
Surprisingly for a lectin, the ability of CD23 to bind IgE does not involve binding to carbohydrate30. However, in addition to IgE, a second CD23 binding ligand (although evolutionarily, this was probably the primary binding partner for CD23) that does bind CD23 in a carbohydrate-dependent manner has been subsequently identified as CD21 (Refs 31, 32). As CD21 is expressed by B cells, follicular dendritic cells (FDCs), activated T cells and basophils, adhesion pairing between these two molecules may have important consequences for allergy. Furthermore, the ability of CD23 to bind both IgE and CD21 links the IgE antibody and complement systems, with consequences for IgE regulation, as described later.
The structure of the CD23 monomeric lectin domain has been determined by both NMR (nuclear magnetic resonance) and X-ray crystallographic analysis27, 33, and although both analyses confirm the C-type lectin fold, the structures differ in the calcium-binding regions (Fig. 4b). Although CD23 appears to retain the two sites that are found in most other members of the family (on the basis of sequence homology), the NMR structure shows calcium occupancy at one site, whereas the X-ray structure shows calcium bound only at the other. Differences in the structures elsewhere point to a domain with a considerable degree of plasticity. The NMR analysis also located the binding sites for both IgE (using monomeric C3) and CD21 (N-terminal domains 1 and 2)27. The isolated C3 domain was used because IgE-Fc containing both C3 domains formed complexes of very high molecular weight27. The CD21 site is incompletely represented in the NMR and crystal structures; the full C-terminal tail region was not present in the construct used in the NMR study, and although it was present in that used for the X-ray analysis, it was not visible in the structure, implying flexibility of this region. However, the IgE and CD21 binding sites are clearly distant from each other (Fig. 4b), consistent with earlier evidence from site-directed mutagenesis34, 35. Further studies showed that both could bind simultaneously27. A model for trimerization of the heads was also proposed based on the NMR data, which brings the three CD21 binding sites close together adjacent to the stalk and leaves the three IgE binding sites positioned around the periphery of the trimer of heads.
3) and CD21 (N-terminal domains 1 and 2)27. The isolated C3 domain was used because IgE-Fc containing both C3 domains formed complexes of very high molecular weight27. The CD21 site is incompletely represented in the NMR and crystal structures; the full C-terminal tail region was not present in the construct used in the NMR study, and although it was present in that used for the X-ray analysis, it was not visible in the structure, implying flexibility of this region. However, the IgE and CD21 binding sites are clearly distant from each other (Fig. 4b), consistent with earlier evidence from site-directed mutagenesis34, 35. Further studies showed that both could bind simultaneously27. A model for trimerization of the heads was also proposed based on the NMR data, which brings the three CD21 binding sites close together adjacent to the stalk and leaves the three IgE binding sites positioned around the periphery of the trimer of heads.
However, the interaction of CD23 with CD21 involves not only domains 1 and 2 of CD21, but also domains 5–8, which interact with CD23 in a carbohydrate-dependent manner32. CD21 consists of a tandem array of 15 or 16 short consensus repeats, a domain with a 6-stranded -barrel structure that is a constituent of many complement-related proteins. The crystal structure of domains 1 and 2 of CD21 is known36, and the whole molecule adopts an extended, flexible structure that is approximately 40 nm long37 (Fig. 5a). CD21 could clearly make extensive, multi-point attachments to the CD23 trimer, perhaps to the head as well as to the tail. However, we are still far from understanding the details of this interaction.
-barrel structure that is a constituent of many complement-related proteins. The crystal structure of domains 1 and 2 of CD21 is known36, and the whole molecule adopts an extended, flexible structure that is approximately 40 nm long37 (Fig. 5a). CD21 could clearly make extensive, multi-point attachments to the CD23 trimer, perhaps to the head as well as to the tail. However, we are still far from understanding the details of this interaction.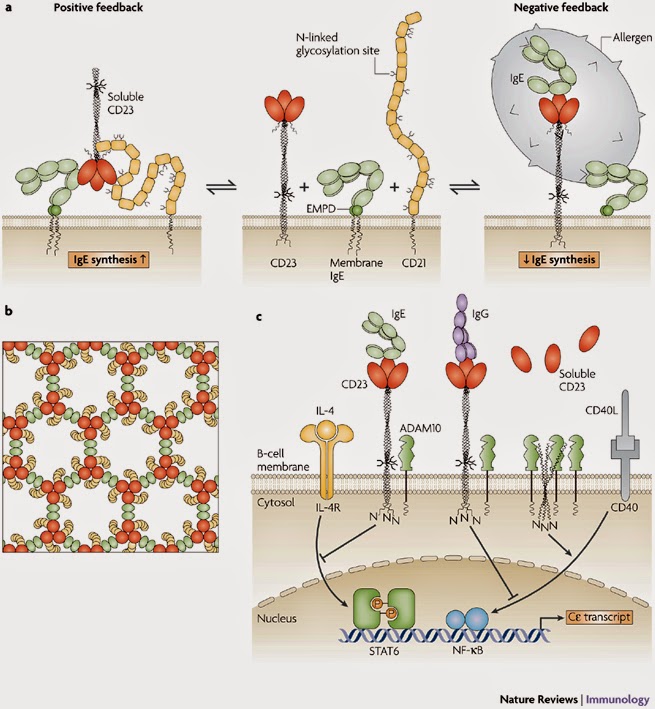 |
| FIGURE 5 | Mechanisms of IgE regulation by CD23.
a | Positive and negative regulation of IgE synthesis by human CD23. In this model, positive regulation of IgE synthesis is a result of the co-ligation of membrane IgE and CD21 on a human B cell committed to IgE synthesis by soluble CD23 released from membrane-bound CD23. IgE synthesis is thereby upregulated via synergistic signalling between the membrane IgE–Ig
–Ig complex and the CD21–CD19 complex. (Soluble CD23 trimerizes in a concentration-dependent manner and on IgE binding, as shown here.) Negative regulation of IgE synthesis occurs by the co-ligation of IgE and CD23 on the membrane by allergen–IgE complexes. Competition between CD21 and CD23 for membrane IgE thus leads to homeostasis. b | A possible model for the formation of a signalling platform involving co-ligation of membrane IgE and CD21 by soluble CD23. It is known that: monomeric soluble CD23 binds simultaneously to a single C3 domain of IgE and one molecule of CD21; IgE-Fc can bind two molecules of monomeric soluble CD23; and soluble CD23 is trimeric at the high concentrations that probably exist in tissues in allergic inflammation and on binding to IgE-Fc. Thus, trimeric soluble CD23 (shown in red) can bind three molecules of IgE (via the C3 domain; shown in green) and three molecules of CD21 (shown in yellow). Furthermore, IgE can link two molecules of soluble trimeric CD23. This may lead to the formation of an extended network, creating a signalling platform for the survival and differentiation of IgE-committed B cells. c | In this scheme, IgE binding to mouse membrane CD23 trimers (which lack the C-terminal tails that in human CD23 trimers bind CD21) interferes with the upregulation of IgE class switching induced by interleukin-4 (IL-4) and CD40 ligand (CD40L). As CD23 trimers are unstable in the B-cell membrane, their unravelling allows ADAM10 (a disintegrin and metalloproteinase 10) to cleave the stalk region, releasing soluble CD23. This cleavage of membrane CD23 allows IgE synthesis to proceed by default. The binding of IgE, or certain CD23-specific IgG antibodies that bind to the lectin head domain (such as lumiliximab), stabilize the trimer and thereby enhance negative signalling. Although the negative signalling pathway of CD23 is unknown, IL-4 and CD40L act through STAT6 (signal transducer and activator of transcription 6) and NF- B (nuclear factor-B), respectively, to stimulate C germline gene transcription and class switching to IgE. EMPD, extracellular membrane-proximal domain; IL-4R, IL-4 receptor. Modified with permission from Ref. 85 © (2007) Current Science Inc. B (nuclear factor-B), respectively, to stimulate C germline gene transcription and class switching to IgE. EMPD, extracellular membrane-proximal domain; IL-4R, IL-4 receptor. Modified with permission from Ref. 85 © (2007) Current Science Inc. |
The binding site for CD23 in IgE-Fc is not precisely mapped either, but lies in the C3 domains distinct from the FcRI site38, away from the local symmetry axis of the Fc, and consistent with the observed 2:1 stoichiometry of binding25. The ability of IgE to engage two molecules of CD23, and the dual binding activity of CD23, suggests a model for the formation of extensive arrays in the cell membrane involving co-crosslinking of membrane IgE and CD21 by trimeric soluble CD23 (Ref. 27). The significance of the equilibrium shown in Fig. 5a, and the extended membrane complex (schematically indicated for one possible array in Fig. 5b) are discussed below.
3 domains distinct from the FcRI site38, away from the local symmetry axis of the Fc, and consistent with the observed 2:1 stoichiometry of binding25. The ability of IgE to engage two molecules of CD23, and the dual binding activity of CD23, suggests a model for the formation of extensive arrays in the cell membrane involving co-crosslinking of membrane IgE and CD21 by trimeric soluble CD23 (Ref. 27). The significance of the equilibrium shown in Fig. 5a, and the extended membrane complex (schematically indicated for one possible array in Fig. 5b) are discussed below.
The wider network. The IgE network extends beyond the two principal receptors. A protein known originally as -binding protein, but now known as galectin-3, has the ability to bind to both IgE and FcRI via -galactose-containing oligosaccharide chains, thereby activating mast cells or basophils by crosslinking receptor-bound IgE, FcRI or both39. Galectin-3 consists of a single carbohydrate-recognition domain (CRD) with an N-terminal region of proline-, glycine- and tyrosine-rich repeats through which it readily forms pentamers in complex with oligosaccharides40; it therefore has a unique architecture and oligomerization propensity within the family of galectins, which otherwise contain just one or two CRDs and most commonly form dimers. The crystal structure of the CRD displays the canonical fold of this family, namely a sandwich of 5- and 6-stranded -sheets, with the oligosaccharide binding site lying within a shallow surface groove41.
-binding protein, but now known as galectin-3, has the ability to bind to both IgE and FcRI via -galactose-containing oligosaccharide chains, thereby activating mast cells or basophils by crosslinking receptor-bound IgE, FcRI or both39. Galectin-3 consists of a single carbohydrate-recognition domain (CRD) with an N-terminal region of proline-, glycine- and tyrosine-rich repeats through which it readily forms pentamers in complex with oligosaccharides40; it therefore has a unique architecture and oligomerization propensity within the family of galectins, which otherwise contain just one or two CRDs and most commonly form dimers. The crystal structure of the CRD displays the canonical fold of this family, namely a sandwich of 5- and 6-stranded -sheets, with the oligosaccharide binding site lying within a shallow surface groove41.
Similarly, CD23 has been shown to interact with several integrins, including M2-integrin in humans42 and mice43, X2-integrin42, V3-integrin44 and, most recently, V5-integrin45. The region of CD23 involved in the interaction with V5-integrin has been located, and is distinct from the IgE and CD21 binding sites but close to the stalk region (Fig. 4b). It includes two adjacent basic residues in a tripeptide motif, Arg-Lys-Cys, located on an exposed disulphide-bonded loop, and is recognized by a site on the integrin -chain that is distinct from the RGD sequence binding site of the integrin45. No further details of these interactions are known.
M2-integrin in humans42 and mice43, X2-integrin42, V3-integrin44 and, most recently, V5-integrin45. The region of CD23 involved in the interaction with V5-integrin has been located, and is distinct from the IgE and CD21 binding sites but close to the stalk region (Fig. 4b). It includes two adjacent basic residues in a tripeptide motif, Arg-Lys-Cys, located on an exposed disulphide-bonded loop, and is recognized by a site on the integrin -chain that is distinct from the RGD sequence binding site of the integrin45. No further details of these interactions are known.Functions of IgE
We now consider the essential features of IgE function involving interactions between members of the protein network and the role of the different cells that express these proteins.
Immediate hypersensitivity: mast cells and basophils. Both IgE and mast cells are concentrated in the mucosal tissues, and so IgE antibodies are among the first defence molecules that an invading pathogen will encounter. The hallmark of an allergic response, which is mediated by the IgE–FcRI complex on mast cells, is immediate hypersensitivity. This reveals itself in characteristic signs and symptoms in the different target organs of allergy, the skin (atopic dermatitis or eczema), the nose (rhinitis), the lungs (asthma) and the gut (food allergic reactions)4, 15. Crosslinking of IgE–FcRI complexes on the mast-cell surface by allergens leads, within minutes, to the so-called 'early phase' of the allergic reaction, which involves mast-cell degranulation and the synthesis of lipid mediators. Cytokines and chemokines liberated in this early phase initiate the 'late phase', which peaks some hours later and involves the recruitment and activation of inflammatory cells at sites sensitive to allergen. Similarly, but without overt symptoms, allergens activate the IgE-sensitized APCs, which in turn promote IgE production by B cells to replenish the IgE consumed in the allergic reaction, thereby maintaining mast-cell and APC sensitization (Fig. 6).
RI complex on mast cells, is immediate hypersensitivity. This reveals itself in characteristic signs and symptoms in the different target organs of allergy, the skin (atopic dermatitis or eczema), the nose (rhinitis), the lungs (asthma) and the gut (food allergic reactions)4, 15. Crosslinking of IgE–FcRI complexes on the mast-cell surface by allergens leads, within minutes, to the so-called 'early phase' of the allergic reaction, which involves mast-cell degranulation and the synthesis of lipid mediators. Cytokines and chemokines liberated in this early phase initiate the 'late phase', which peaks some hours later and involves the recruitment and activation of inflammatory cells at sites sensitive to allergen. Similarly, but without overt symptoms, allergens activate the IgE-sensitized APCs, which in turn promote IgE production by B cells to replenish the IgE consumed in the allergic reaction, thereby maintaining mast-cell and APC sensitization (Fig. 6). |
| FIGURE 6 | Pump priming of the allergic response by allergens.
IgE is synthesized and secreted by B cells that have undergone heavy-chain class switching from IgM to IgE. The IgE binds to Fc
RI on mast cells and antigen-presenting cells (APCs) (a) and sensitizes these cells to allergens. Omalizumab inhibits the binding of IgE to FcRI on mast cells and APCs (b). Allergen binding to IgE triggers mast-cell degranulation to cause an allergic response (c). Allergen binding to the APC leads to the presentation of allergenic peptides to T helper 2 (TH2) cells (d). The allergen-activated TH2 cells secrete interleukin-4 (IL-4) (e) to maintain the TH2-cell lineage and recruit more TH cells into this lineage (e). The TH2 cells also secrete IL-13 and express CD40 ligand (CD40L), which together with IL-4 stimulates heavy-chain class switching to IgE (f). The allergen-activated mast cells contribute to the production of IL-4 and IL-13 (and express CD40L), which may also stimulate heavy-chain class switching to IgE (g). IL-4, IL-13 and CD40L also stimulate the expression of CD23 and the release of soluble CD23 (h). In humans, soluble trimeric CD23 upregulates IgE synthesis and secretion through interaction with CD21 (i). Thus, allergen acts in pump priming of the allergic response. |
The processes of mast-cell and APC recruitment and IgE production in the mucosal tissues are central to the functions of IgE, and no less so in guarding against systemic anaphylaxis. Mast-cell precursors are generated in the bone marrow and migrate to mucosal tissue before expressing FcRI (Ref. 46). The receptor is highly expressed ( 500,000 copies per cell) on tissue mast cells, probably as a result of IgE-mediated upregulation of FcRI expression15, 47. The concentrations of IgE required for the upregulation of FcRI in the mucosal tissues, which are higher than those normally present in the circulation, may derive from IgE synthesis by local B cells48. The rate of this process is more than sufficient to maintain saturation of the FcRI molecules on the mast cells4, 48, and any excess IgE is preferentially directed into secretions, rather than entering the circulation49, 50. Local IgE synthesis is important for the function of IgE, as is discussed later.
500,000 copies per cell) on tissue mast cells, probably as a result of IgE-mediated upregulation of FcRI expression15, 47. The concentrations of IgE required for the upregulation of FcRI in the mucosal tissues, which are higher than those normally present in the circulation, may derive from IgE synthesis by local B cells48. The rate of this process is more than sufficient to maintain saturation of the FcRI molecules on the mast cells4, 48, and any excess IgE is preferentially directed into secretions, rather than entering the circulation49, 50. Local IgE synthesis is important for the function of IgE, as is discussed later.
RI (Ref. 46). The receptor is highly expressed (500,000 copies per cell) on tissue mast cells, probably as a result of IgE-mediated upregulation of FcRI expression15, 47. The concentrations of IgE required for the upregulation of FcRI in the mucosal tissues, which are higher than those normally present in the circulation, may derive from IgE synthesis by local B cells48. The rate of this process is more than sufficient to maintain saturation of the FcRI molecules on the mast cells4, 48, and any excess IgE is preferentially directed into secretions, rather than entering the circulation49, 50. Local IgE synthesis is important for the function of IgE, as is discussed later.
Cytokinergic IgE activation of mast cells. Recent work has revealed that certain monomeric IgE molecules have the ability to activate mast-cell signalling in the absence of crosslinking by allergen through some of the same pathways as allergen–IgE complexes. The reported activities vary from the most 'highly cytokinergic', corresponding to full mast-cell activation (degranulation) at one extreme to the most 'poorly cytokinergic' (enhanced mast-cell survival) at the other. Although cytokinergic IgE molecules were shown to be monomeric in solution, on the cell membrane they form clusters and thereby crosslink FcRI and activate mast-cell signalling. The effective concentrations of IgE on the cell surface exceed those in solution by many orders of magnitude, and their orientation may further promote self association. The structural determinants of cytokinergic activity are under investigation. It is not yet clear whether these IgE molecules act in the same manner in vivo, and if so what the physiological consequences might be. This subject is discussed in greater detail in two recent reviews15, 51.
RI and activate mast-cell signalling. The effective concentrations of IgE on the cell surface exceed those in solution by many orders of magnitude, and their orientation may further promote self association. The structural determinants of cytokinergic activity are under investigation. It is not yet clear whether these IgE molecules act in the same manner in vivo, and if so what the physiological consequences might be. This subject is discussed in greater detail in two recent reviews15, 51.
Local IgE synthesis: B cells and plasma cells. In allergy and asthma, the populations of B cells and plasma cells in the respiratory tract mucosa are heavily biased towards the production of IgE. Approximately 4% of the B cells and 12–19% of the plasma cells express IgE in the nasal mucosa of patients with rhinitis, compared with 1% and <1 healthy="" in="" respectively="" subjects="" sup="">52
The expression of IgE requires class-switch recombination (CSR) of the variable (VDJ) region, initially linked to the constant (C) region of another antibody class (C , C or C), to C, in the immunoglobulin heavy-chain locus. CSR to IgE is induced by the cytokines IL-4 or IL-13 secreted by T helper 2 (TH2) cells. Until recently, CSR had been thought to occur only in the germinal centres of lymphoid tissue. The origin of the IgE-expressing B cells and plasma cells in the respiratory tract mucosa was revealed by the observation of CSR in these B cells54, 55, 56, 57, 58, 59. Moreover, somatic hypermutation (SHM) and clonal expansion of respiratory-tract B cells was observed, implying that clonal selection and affinity maturation operate in the B-cell population of the tissue54, 55, 58. The conditions of allergic inflammation may therefore shape the local antibody repertoire, favouring the production of high-affinity IgE antibodies. The synthesis of IgE directed against grass pollen allergens was found to persist beyond the grass pollen season48; this is consistent with the hypothesis that inflamed tissues, as well as the bone marrow, constitute a survival niche for antibody-secreting plasma cells60, 61.
, C or C), to C, in the immunoglobulin heavy-chain locus. CSR to IgE is induced by the cytokines IL-4 or IL-13 secreted by T helper 2 (TH2) cells. Until recently, CSR had been thought to occur only in the germinal centres of lymphoid tissue. The origin of the IgE-expressing B cells and plasma cells in the respiratory tract mucosa was revealed by the observation of CSR in these B cells54, 55, 56, 57, 58, 59. Moreover, somatic hypermutation (SHM) and clonal expansion of respiratory-tract B cells was observed, implying that clonal selection and affinity maturation operate in the B-cell population of the tissue54, 55, 58. The conditions of allergic inflammation may therefore shape the local antibody repertoire, favouring the production of high-affinity IgE antibodies. The synthesis of IgE directed against grass pollen allergens was found to persist beyond the grass pollen season48; this is consistent with the hypothesis that inflamed tissues, as well as the bone marrow, constitute a survival niche for antibody-secreting plasma cells60, 61.
, C or C), to C, in the immunoglobulin heavy-chain locus. CSR to IgE is induced by the cytokines IL-4 or IL-13 secreted by T helper 2 (TH2) cells. Until recently, CSR had been thought to occur only in the germinal centres of lymphoid tissue. The origin of the IgE-expressing B cells and plasma cells in the respiratory tract mucosa was revealed by the observation of CSR in these B cells54, 55, 56, 57, 58, 59. Moreover, somatic hypermutation (SHM) and clonal expansion of respiratory-tract B cells was observed, implying that clonal selection and affinity maturation operate in the B-cell population of the tissue54, 55, 58. The conditions of allergic inflammation may therefore shape the local antibody repertoire, favouring the production of high-affinity IgE antibodies. The synthesis of IgE directed against grass pollen allergens was found to persist beyond the grass pollen season48; this is consistent with the hypothesis that inflamed tissues, as well as the bone marrow, constitute a survival niche for antibody-secreting plasma cells60, 61.
Germinal-centre-like reactions and antibody synthesis are not confined to the respiratory tract of atopic individuals or to IgE. Indeed, both local CSR to IgE and IgE synthesis also occur in the bronchial mucosa of non-atopic asthmatics59, although the specificity of the IgE in these individuals is unknown. The gastrointestinal mucosa is an abundant source of both IgA and IgE, and can support CSR to both these isotypes62, 63. CSR to IgA is favoured in the gastrointestinal mucosa, an environment seen to be under constant antigenic challenge, although it is generally programmed or educated to tolerate foods and commensal bacteria64, 65. However, IgE is expressed more abundantly in the gastrointestinal tract of individuals with food allergies than in those without66. A deeper understanding of the mechanisms involved in oral tolerance may suggest ways of diverting the immune response away from IgE production in allergic individuals.
Allergen presentation: Langerhans cells, dendritic cells and B cells. IgE binds to FcRI on the surface of Langerhans cells and to the interdigitating epithelial dendritic cells, which are all engaged at our body surfaces in the surveillance for antigens in the environment. Following the capture of allergens they either migrate to local lymph nodes or possibly remain in the tissues where they present the processed allergens to TH cells67. Similarly, allergen–IgE complexes bound in the mucosa to CD23 that is expressed by allergen-activated B cells can facilitate antigen presentation to the T cells68. The process of antigen presentation by way of CD23 is termed facilitated antigen presentation (FAP). The interaction between CD23 and HLA-DR in the cell membrane is involved in the trafficking of allergen–IgE–CD23 complexes to endosomes, where the allergen-derived peptides are loaded onto the HLA-DR molecules for presentation at the B-cell surface69.
RI on the surface of Langerhans cells and to the interdigitating epithelial dendritic cells, which are all engaged at our body surfaces in the surveillance for antigens in the environment. Following the capture of allergens they either migrate to local lymph nodes or possibly remain in the tissues where they present the processed allergens to TH cells67. Similarly, allergen–IgE complexes bound in the mucosa to CD23 that is expressed by allergen-activated B cells can facilitate antigen presentation to the T cells68. The process of antigen presentation by way of CD23 is termed facilitated antigen presentation (FAP). The interaction between CD23 and HLA-DR in the cell membrane is involved in the trafficking of allergen–IgE–CD23 complexes to endosomes, where the allergen-derived peptides are loaded onto the HLA-DR molecules for presentation at the B-cell surface69.
Antigen presentation through the membrane-bound B-cell receptor (BCR) involves the interaction of cognate B cells with TH cells; this is therefore limited by the number of cognate B cells68. However, antigen presentation through secreted IgE via FAP may overcome this limitation as all antigen-activated (that is, CD23-expressing) B cells are able to present a variety of peptides, even from totally unrelated allergens, to cognate T cells, regardless of the specificity of the B cell's own antigen receptor. This is a probable scenario for 'epitope spreading'70, not just within a single allergen (Fig. 7a), but to unrelated allergens (Fig. 7b). An antigen-activated B cell expressing CD23 can in effect behave as a 'professional' APC such as a dendritic cell, which can simultaneously process unrelated antigens through FcRs and cause epitope spreading to other antigens. Indeed, CD23-mediated FAP is known to be as efficient as FcR-mediated antigen presentation by dendritic cells70, orders of magnitude more efficient than B-cell internalization via surface immunoglobulin (the BCR). Furthermore, whereas FAP enhances the presentation of allergens recognized by pre-existing IgE molecules, it suppresses that of other antigens68. FAP therefore serves in a positive-feedback mechanism to enhance allergic sensitization (Fig. 7b). This is a clinically serious phenomenon, as monospecific allergies often deteriorate into polyspecific allergies.
Rs and cause epitope spreading to other antigens. Indeed, CD23-mediated FAP is known to be as efficient as FcR-mediated antigen presentation by dendritic cells70, orders of magnitude more efficient than B-cell internalization via surface immunoglobulin (the BCR). Furthermore, whereas FAP enhances the presentation of allergens recognized by pre-existing IgE molecules, it suppresses that of other antigens68. FAP therefore serves in a positive-feedback mechanism to enhance allergic sensitization (Fig. 7b). This is a clinically serious phenomenon, as monospecific allergies often deteriorate into polyspecific allergies. |
| FIGURE 7 | CD23-dependent epitope spreading.
a | Antigen X, recognized by a B-cell receptor (BCR) specific for epitope 1, is internalized, processed and presented by MHC class II molecules. T cells specific for antigen X1 participate in cognate interactions with the B cell, leading to the production of IgG antibodies specific for epitope X1, and, if followed by class-switch recombination (CSR), IgE antibodies. T cells generated in this initial process, specific for other peptide fragments (X2-specific T cells), may take part in cognate interactions with B cells specific for other epitopes (X2-specific B cells), leading to IgG and perhaps also IgE specific for epitope X2. Thus, B cells acting as antigen-presenting cells (APCs) using surface immunoglobulin 'spread' the response from one epitope to another, but only within the same antigen. b | By contrast, if the B cell that is activated by antigen expresses membrane CD23, it can internalize IgE bound to an allergen (Y) that is unrelated to antigen X and the specificity of that B cell. The figure shows a cognate interaction that leads to IgG and IgE specific for epitope X2 (as in part a), but the CD23-mediated internalization of allergen Y, to which a low titre of specific IgE antibodies must initially exist, leads to the production of allergen-Y-specific IgG and then IgE antibodies. The response thus spreads from antigen X to the unrelated allergen Y. Internalization of other allergen–IgE complexes by CD23 can further spread the response, just as 'professional' APCs bearing Fc receptors for IgG (Fc
Rs) simultaneously process unrelated antigens and spread IgG responses from one antigen to another. CD23-mediated antigen presentation is more efficient than surface-immunoglobulin-mediated antigen presentation, and therefore can enhance a low level response to an allergen (such as allergen Y) and promote spreading to other allergens. This epitope spreading from one allergen to another may explain how certain allergic subjects deteriorate from sensitivity to only one allergen to multiple sensitivities70. |
Recently discovered polymorphisms in the CD23 gene may relate to the phenomena of CD23-mediated FAP and epitope spreading. Mutations in the stalk region of murine CD23 are associated with destabilization of the stalk, decreased expression on the cell membrane and elevated CSR to IgE71, 72. The Arg62Trp polymorphism in human CD23 occurs at the base of the CD23 stalk region and confers resistance to enzymatic cleavage73; it also lies within a peptide sequence that has been shown to affect the interaction between MHC class II molecules and CD23 (see earlier)26. It would therefore be interesting to examine the effects of this polymorphism on FAP and the ability of CD23-expressing B cells to promote epitope spreading. Earlier functional studies have focused only on polymorphisms in the genes that encode the FcRI -chain and -chain3.
RI -chain and -chain3.
Other effector functions of IgE and CD23: human monocytes and eosinophils. Monocytes and eosinophils are among the different IgE effector cells that are drawn to sites of allergic inflammation. IgE upregulates the expression of FcRI, whereas IL-4 and IL-13 stimulate the expression of CD23 by these cells. This in turn arms the cells for useful functions, such as clearance of antigen–IgE complexes, and killing and phagocytosis of pathogens (for example, helminth parasites)74 and tumour cells that bear 'foreign' antigens (for example, re-expressed neonatal antigens)75. The ability of FcRI and CD23 to mediate IgE-dependent monocyte- and eosinophil-mediated lysis of target cells and phagocytosis of the resulting cell fragments was recently shown for the first time by experiments with an IgE antibody against tumour cells75.
RI, whereas IL-4 and IL-13 stimulate the expression of CD23 by these cells. This in turn arms the cells for useful functions, such as clearance of antigen–IgE complexes, and killing and phagocytosis of pathogens (for example, helminth parasites)74 and tumour cells that bear 'foreign' antigens (for example, re-expressed neonatal antigens)75. The ability of FcRI and CD23 to mediate IgE-dependent monocyte- and eosinophil-mediated lysis of target cells and phagocytosis of the resulting cell fragments was recently shown for the first time by experiments with an IgE antibody against tumour cells75.
The upregulation of CD23 also leads to the release of soluble fragments implicated in IgE-independent monocyte-mediated cytotoxicity. The CD23 fragments bind to M2-integrin and X2-integrin on monocytes, and thereby initiate the production of tumour-necrosis factor and other pro-inflammatory cytokines (such as IL-1 and IL-6)76. Indeed, CD23 fragments and membrane-associated CD23 probably act sequentially in monocyte-mediated cell killing, the former acting first to mediate cell lysis, the latter disposing of the fragments. In carrying out their functions, monocytes and eosinophils inevitably also inflict some damage on bystander cells in the tissue. This side effect ultimately contributes to tissue remodelling and exacerbates the symptoms of chronic asthma, and to the compromised function of IgE in the defence against parasites74.
M2-integrin and X2-integrin on monocytes, and thereby initiate the production of tumour-necrosis factor and other pro-inflammatory cytokines (such as IL-1 and IL-6)76. Indeed, CD23 fragments and membrane-associated CD23 probably act sequentially in monocyte-mediated cell killing, the former acting first to mediate cell lysis, the latter disposing of the fragments. In carrying out their functions, monocytes and eosinophils inevitably also inflict some damage on bystander cells in the tissue. This side effect ultimately contributes to tissue remodelling and exacerbates the symptoms of chronic asthma, and to the compromised function of IgE in the defence against parasites74.
The IgE-binding protein galectin-3, which is expressed by monocytes, has both IgE-dependent and IgE-independent pro-inflammatory activities in the allergic response77. Its expression is elevated in peribronchial monocytes and it is released into the extracellular space. It contributes to cell survival and activation and to the retention of cells in the extracellular matrix in allergic inflammation. Galectin-3 also enhances the hypersensitivity of mast cells and the phagocytic activity of monocytes77, 78, perhaps by crosslinking additional IgE and FcRI molecules on these cells when they have engaged their specific targets. The pentameric molecule galectin-3 may of itself contribute through FcRI to airway smooth-muscle-cell contractile responses, and thus to airway hyper-responsiveness and remodelling in asthma.
RI molecules on these cells when they have engaged their specific targets. The pentameric molecule galectin-3 may of itself contribute through FcRI to airway smooth-muscle-cell contractile responses, and thus to airway hyper-responsiveness and remodelling in asthma.
IgE transport through the gastrointestinal epithelium: epithelial cells. CD23 is expressed (as by many other immune cells) by rodent and human intestinal epithelial cells79. Recent data indicate that CD23 on epithelial cells plays a central part in transporting IgE and allergen–IgE complexes across the epithelial-cell barrier and into the underlying tissue, bypassing the tight junctions between the epithelial cells (Fig. 8). Such a mechanism is used to take up maternal IgE in amniotic fluid swallowed by the fetus2. IgE is also synthesized locally in the gut and secreted into the gut lumen80, 81. There, the secreted IgE binds to allergens and the resulting complexes are delivered intact to mast cells in the gastrointestinal mucosa to trigger food allergic reactions. In this sense, CD23 resembles the neonatal Fc receptor for IgG (FcRn) and the polyimmunoglobulin receptor (pIgR) for IgA, which both facilitate transcytosis of immunoglobulins. The release of pro-inflammatory mediators by the activated mast cells, epithelial cells and other cells, all attracted to the site of allergic inflammation, eventually causes a nonspecific reduction in the barrier function of the epithelium and exacerbation of the disease process82. FcRI- and CD23-expressing APCs may also capture the allergen–IgE complexes in the gut and cause the resident TH2 cells to enhance synthesis of allergen-specific IgE in the B cells (Fig. 8).
RI- and CD23-expressing APCs may also capture the allergen–IgE complexes in the gut and cause the resident TH2 cells to enhance synthesis of allergen-specific IgE in the B cells (Fig. 8). |
| FIGURE 8 | Role of CD23 on epithelial cells in the pathogenesis of food allergic disease.
a | Allergen–IgE complexes are captured by CD23 on the luminal side of the intestinal epithelium and transported into the mucosa, where they bind to Fc
RI on mast cells and dendritic cells (DCs), causing allergic inflammation and local IgE class switching in B cells and IgE synthesis (1). This occurs in the fetus by the swallowing of amniotic fluid containing maternal allergen–IgE complexes, and may also represent the mechanism of early sensitization to food allergens in the adult intestinal tract. b | IgE synthesized by plasma cells in the mucosa are transported by CD23 on the mucosal side of the epithelium into the gut lumen, where they capture allergens and bind to CD23 on the luminal side of the epithelium (2) to be delivered to the mucosa. c | Finally, the inflammation caused by allergic reactions mediated by IgE in the mucosa damages the intestinal epithelium, disrupting the tight junctions between the epithelial cells. Free allergens can now pass between the cells and bind to the IgE-sensitized mast cells and DCs in the mucosa, exacerbating the food allergy (3). |
This may explain how sensitization to food comes about, in contrast to oral tolerance, which is the usual outcome of the encounter of a protein in the gastrointestinal tract64, 65. There is evidence for local synthesis of IgE concomitantly with CD23 expression in the fetus2, 83; parental history of atopy, and titre of cord-blood IgE, are predictive of early atopy84. Thus, the bidirectional transport of IgE in the immature gastrointestinal tract, mediated by CD23, may have an important bearing on early sensitization to allergens, preceding the appearance of atopy (Fig. 9). The effects of polymorphisms in the CD23 gene on the development of atopy in mice and humans are fully consistent with this hypothesis85. Once established, atopy may endure for many years and is self-perpetuating. Epitope spreading exacerbates the problem, with the establishment of a positive-feedback loop culminating in indiscriminate sensitization to unrelated allergens.
 |
| FIGURE 9 | Allergic sensitization and positive feedback.
The interaction of genes and environmental factors determine the risk of allergic sensitization3. By early 2006, six genes had been identified by positional cloning and over 100 by candidate gene association98, 99. This figure shows only some of the genes in which polymorphisms are directly involved in the synthesis or effector functions of IgE. Equally, there are many environmental factors associated with allergy. These include the level of exposure to allergens100; the way in which food is processed101, 102; elevated pH of the stomach contents of the fetus and young babies, or caused by antacid medications in adults103; atmospheric pollution104; respiratory syncytial virus and rhinovirus infections during infancy105, 106; Staphylococcal aureus infections, which generate superallergenic enterotoxins107, 108; and the activity of autoantibodies109, 110. Each of these factors might have only a small effect, but their interaction is likely to be crucial in the development of atopy. Once sensitization occurs, these factors continue to operate, and atopy is exacerbated by the resulting inflammation, persistent IgE synthesis and epitope spreading. FCERIB, high-affinity Fc receptor for IgE; IL, interleukin; ILR, IL receptor; STAT6, signal transducer and activator of transcription 6.
|
IgE homeostasis
As indicated above, both IgE synthesis and its effector functions appear to be largely confined to the mucosa. The concentrations of IgE in the circulation are maintained at a low level, normally at approximately 0.1 g per ml compared to 10 mg per ml for IgG, and may only be increased by about 10-fold in allergic individuals. This argues for the existence of mechanisms that regulate the anatomical distribution of IgE for optimal function and for the health of the individual (and perhaps even survival of the species), by suppressing its potential as a mediator of systemic anaphylaxis. Here, we consider recent evidence concerning the molecular mechanisms of IgE regulation through the network of its interacting proteins.
g per ml compared to 10 mg per ml for IgG, and may only be increased by about 10-fold in allergic individuals. This argues for the existence of mechanisms that regulate the anatomical distribution of IgE for optimal function and for the health of the individual (and perhaps even survival of the species), by suppressing its potential as a mediator of systemic anaphylaxis. Here, we consider recent evidence concerning the molecular mechanisms of IgE regulation through the network of its interacting proteins.
Positive and negative regulation by CD23. IL-4 and IL-13, the cytokines required for CSR to IgE, also induce the upregulation of CD23 expression by B cells and inflammatory cells (Fig. 6). This CD23 can then act as a buffer against the accumulation of excessive, possibly dangerous, concentrations of free IgE; the FcRI site on the IgE is blocked by bound CD23, thereby rendering the antibody inactive. Nevertheless, IgE can exert its functions through FcRI at a lower concentration range, by reason of its higher affinity for this receptor.
RI site on the IgE is blocked by bound CD23, thereby rendering the antibody inactive. Nevertheless, IgE can exert its functions through FcRI at a lower concentration range, by reason of its higher affinity for this receptor.
In addition to its buffering activity, CD23 has a central role in the regulation of IgE synthesis, although the mechanisms are still debated. There are two current schools of thought85. According to one, CD23 both enhances IgE synthesis at low IgE concentrations and suppresses it at high concentrations, engaging in both positive and negative feedback86 (Fig. 5a). In this view, soluble CD23, cleaved from membrane CD23 that lacks protection by IgE, stimulates upregulation of IgE synthesis by the co-ligation of membrane IgE and CD21. This mechanism is analogous to that by which co-ligation of IgM and CD21 by fragments of complement component C3 attached to antigen stimulates the immune response86, 87. The extended array of membrane IgE and CD21 co-ligated by the soluble CD23 fragments (Fig. 5b) could then act as a signalling platform for the proliferation and differentiation of the B cells into IgE-secreting plasma cells. Protection of membrane CD23 by IgE binding prevents the release of soluble CD23 to abrogate positive signalling for IgE synthesis. In addition, the co-ligation of membrane CD23 and membrane IgE by allergen–IgE complexes results in negative signalling for IgE synthesis.
A slimmer version of this model, proposes that soluble CD23 stimulates IgE synthesis by binding to CD21 alone (without membrane IgE), and IgE alone (without allergen) suppresses IgE synthesis by binding to CD23 alone (without membrane IgE). This would be the human counterpart of the second model. Ockam's razor would favour the slimmer model, were it not for the evidence that CD23 is able to bind simultaneously to IgE and CD21 as illustrated in Fig. 5a, b.
The second model85 also proposes that IgE binding to membrane CD23 results in negative signalling (Fig. 5c), but does not support the positive signalling mediated by soluble CD23 through CD21 that is described above. The negative signalling pathway is consistent with the dramatic upregulation of IgE synthesis in CD23-deficient mice88. However, this does not preclude the existence of a positive signalling pathway in the human system, as soluble CD23 cannot bind to CD21 in mice; mouse CD23 lacks the C-terminal tail, which in human CD23 contains the binding site for the N-terminal domains 1 and 2 of CD21 and is required for the upregulation of IgE synthesis in this system35. It is therefore likely that only a negative-feedback mechanism operates in mice, whereas both positive- and negative-feedback mechanisms operate in the human system. This could be one reason why humans but not mice suffer from allergy.
Regardless of this issue, inhibition of CD23 proteolysis (Fig. 5c) could be a promising therapeutic approach. CD23-specific antibodies do indeed inhibit human IgE synthesis in vitro25, 89, and a primatized CD23-specific antibody, known as lumiliximab (IDEC-152) showed efficacy in a Phase I clinical trial for the treatment of asthma89.
Mechanisms inherent in the immunoglobulin locus. B-cell proliferation is required for CSR and, for reasons still obscure, more cycles of cell division precede the switch to IgE than to IgG90. The use of an alternative pathway of CSR could be one reason91 and sequential switching through other isotypes another57. Chromatin structure may also be an obstacle92, as may sequence-specific factors, such as the position of the germline gene in the immunoglobulin heavy-chain locus, the relatively short length of the 'switch' region (S: 0.8 kb versus S: 1.5 kb), or the DNA sequence of this region. Many double-strand breaks are introduced into the immunoglobulin heavy-chain gene locus during the process of CSR93, and the cells that fail to maintain an intact chromosome during this process undergo apoptosis. Consequently, cell division is often accompanied by cell death and few cells may survive long enough to switch to IgE.
: 0.8 kb versus S: 1.5 kb), or the DNA sequence of this region. Many double-strand breaks are introduced into the immunoglobulin heavy-chain gene locus during the process of CSR93, and the cells that fail to maintain an intact chromosome during this process undergo apoptosis. Consequently, cell division is often accompanied by cell death and few cells may survive long enough to switch to IgE.
After the B cells have switched to a new isotype they must undergo differentiation into plasma cells. This process is also more difficult for the cells that have switched to IgE rather than to other isotypes, owing to a 'fault' of chain termination94. The optimal chain termination signal, AATAAA, which is present in all other isotypes, is altered to AGTAAA, AAGAA and ATTAAA, at the 3' end of the IgE locus, affecting only the expression of the membrane form of IgE. As with other isotypes, signalling through membrane IgE is inseparable from cell survival, and therefore failure of IgE to reach the cell surface results in a high frequency of cell death.
An additional factor is that even when IgE has reached the cell surface, its orientation, in particular the position of the Fab arms, may be less favourable for antigen binding than the secreted IgE in its complex with FcRI (Fig. 3b, c). The bent structure of IgE may, for membrane-bound IgE, limit the possibilities for antigen binding, although the long form of IgE, with its larger EMPD (Fig. 5a) and perhaps greater conformational freedom, may have an advantage over the short membrane-tethered form in this respect12. It has been shown that crosslinking of the two isoforms leads to different kinetics of protein tyrosine phosphorylation and the ensuing cellular responses: crosslinking of the short form leads to inhibition of B-cell growth, whereas signalling by the long form is permissive for cell proliferation12.
RI (Fig. 3b, c). The bent structure of IgE may, for membrane-bound IgE, limit the possibilities for antigen binding, although the long form of IgE, with its larger EMPD (Fig. 5a) and perhaps greater conformational freedom, may have an advantage over the short membrane-tethered form in this respect12. It has been shown that crosslinking of the two isoforms leads to different kinetics of protein tyrosine phosphorylation and the ensuing cellular responses: crosslinking of the short form leads to inhibition of B-cell growth, whereas signalling by the long form is permissive for cell proliferation12.
Although the 'road to IgE production is long and winding'95, it presents manifold opportunities for intervention in the allergic response, but we need first to identify the signposts along this road.
Prevention and treatment of allergic disease
More research has hitherto been devoted to therapy rather than prevention. The concept of a non-anaphylactogenic IgE-specific antibody — one that binds to a site in IgE required for binding to FcRI — is not new4, but has been slow to reach the clinic in the form of omalizumab (Fig. 6). This antibody is proving its value in an ever-widening range of allergic disorders96, but the expense of producing it is prohibitive for all but a small range of applications; it is currently approved for use only in moderate to severe asthma, and then only in cases already found refractory to other (less costly) treatments. Therefore, an attractive option will be the design of small molecule inhibitors of the IgE–FcRI interaction, based on what is known about the structural changes accompanying complex formation, to 'lock' IgE or FcRI in an inactive conformation4, 10, 97.
RI — is not new4, but has been slow to reach the clinic in the form of omalizumab (Fig. 6). This antibody is proving its value in an ever-widening range of allergic disorders96, but the expense of producing it is prohibitive for all but a small range of applications; it is currently approved for use only in moderate to severe asthma, and then only in cases already found refractory to other (less costly) treatments. Therefore, an attractive option will be the design of small molecule inhibitors of the IgE–FcRI interaction, based on what is known about the structural changes accompanying complex formation, to 'lock' IgE or FcRI in an inactive conformation4, 10, 97.
Omalizumab effectively neutralizes IgE, and may even cause apoptosis of committed B cells by crosslinking membrane IgE96. It is unlikely however to affect the long-lived plasma cells that express little of the membrane form of IgE. While the tendency of allergic individuals to mount TH2-cell responses in their target organs persists, there is also the likelihood of relapse if the treatment with the antibody (or a small molecule inhibitor) ceases. The exploratory clinical trials combining immunotherapy, to achieve immune deviation from TH2-type to TH1-type responses, with omalizumab, represent an exciting development.
It is said that an ounce of prevention is worth a pound of cure. Encouragingly, there is growing interest in understanding the sensitization phase of allergic disease, especially in early life (Fig. 9). In this regard, CD23 again appears to be a promising target, for it may, as we have indicated, play a prominent role in the development of atopy. CD23 or IgE could be used first as a biomarker (in blood or stools) and then as a target for therapy in children at risk. Whether lumiliximab, which has shown promise in Phase I clinical trials for asthma, or inhibitors of CD23 signalling pathways, which have proved effective in rodent models of food allergy, or indeed omalizumab, could be safely administered to children is a question for the future.
Links
DATABASES
RCSB Protein Data Bank
1F6A | 1FP5 | 1J87 | 1O0V | 1T8D | 2H2T
Entrez-Gene
FcRI | CD23 | galectin-3 | CD21 | M2-integrin | X2-integrin | ADAM10
Review
Nature Reviews Immunology 8, 205-217 (March 2008) | doi:10.1038/nri2273
Focus on: Allergy and asthma
FURTHER INFORMATION
Allergy and asthma group homepage
Acknowledgements
We acknowledge the support of the Medical Research Council (UK), The Wellcome Trust and Asthma UK for our work in this field. We also thank K. Kirwan, A. Davies and R. Beavil for help with preparation of the original figures.
About the authors
Hannah Gould received her Ph.D. from Harvard University, USA, and carried out postdoctoral research at The National Institute for Medical Research, University College London, UK, and Imperial College London, UK. She joined the Biophysics Department of King's College London, UK, in 1980, and is now Professor of Biophysics and a member of the Medical Research Council (MRC) & Asthma UK Centre in Allergic Mechanisms of Asthma. Her interests in IgE biology include the mechanism of gene regulation at the immunoglobulin loci, the structure and function of proteins of the IgE network, and more recently the application of IgE antibodies to the immunotherapy of cancer.
Brian Sutton received his D.Phil. from the University of Oxford, UK, and continued his postdoctoral research there in the Laboratory of Molecular Biophysics. He joined King's College London in 1987 and began collaborating with Hannah Gould in studies of the IgE network. He is currently Professor of Molecular Biophysics, Head of the Structural Biology Section and a member of the MRC & Asthma UK Centre in Allergic Mechanisms of Asthma. His research focuses on understanding the molecular mechanisms of allergy and asthma, and applying structure-based approaches to develop new therapeutic agents.
Author affiliations
- Randall Division of Cell & Molecular Biophysics, and MRC & Asthma UK Centre in Allergic Mechanisms of Asthma, King's College London, New Hunt's House, Guy's Campus, London, SE1 1UL, UK.
Email: hannah.gould@kcl.ac.uk; Email: brian.sutton@kcl.ac.uk


















0 comments :
Post a Comment